薬理学総論(General Principles)
- manager
- 久野高義
1、ヘルシンキ宣言と臨床試験実施の統一基準(世界医師会、World Medical Association)
1964年:ヘルシンキ宣言:ヒトを対象とするすべての臨床試験の倫理的規範を定める。
1995年:医薬品規制ハーモナイゼーション国際会議(横浜)、日米欧は臨床試験の統一基準の合意。
1998年から新GCPの本格的実施。
1)医薬品の臨床試験の実施の基準(Good Clinical Practice、GCP)、2)文書によるインフォームドコンセント、3)治験審査委員会 (ヘルシンキ宣言をみる)
2、薬の開発過程
薬になる確率は1/30000、10-20年の歳月と500-1000億円以上が必要。
1) 開発目標 |
|
|
|---|---|---|
2) 素材探し | 天然、合成 |
|
3)スクリーニングテスト |
|
|
4) 非臨床試験 |
| |
| a.毒性試験 1.単回投与毒性試験(急性毒性) 2種類以上の動物でLD50 2. 反復投与毒性試験 亜急性(3-7日) 3. 生殖発生毒性試験 4. 遺伝毒性(変異原性)試験 5. 癌原性試験 6. 抗原性試験 7. 局所刺激性試験 | 医薬品の安全性に関する非臨床試験の実施の基準 (Good Laboratory Practice、GLP)
|
| b.薬効薬理試験 1. 薬理作用 ED50、IC50 2. 安全性薬理試験 |
|
| c.薬物動態試験 1. 投与法、吸収 2. 分布 3. 代謝 4. 排泄 |
|
5) 臨床試験
| 二重盲検法 | |
a.第1相試験 | 少数健康男子、安全性、投与量、体内動態 | |
b.第2相試験 | 少数患者、安全性、有効性 | |
c.第3相試験 | 多数の患者、安全性、有効性 | |
6) 申請・審査 | 医薬品医機器総合機構 |
|
7) 生産・販売 |
| |
8) 第4相試験 | 長期安全性と有効性、市販直後調査6ヶ月間 | 市販後医薬品調査実施基準(Good Post-Marketing Surveillance Practice、GPMSP) |
9) 新薬の再審査 | 6~10年目に行う |
|
| 10) 再評価 | 5年間隔で有効性、安全性、品質の見直し |
3、薬物と生体反応
A.薬物の用量と作用:用量-反応曲線
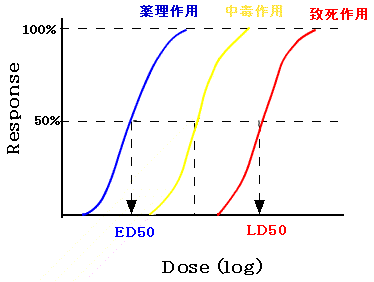
安全域(safety margin)=LD50/ED50。安全域は治療係数(therapeutic index)ともいう。安全係数の大きいものほど、毒性の現れる危険性が少ない。一般に常用量はED50に近い量を投与している。安全域の小さい薬物:ジギタリス(digitalis)、フェニトイン(phenytoin)、炭酸リチウム(lithium carbonate)など。
ヒトでの用量の決め方:感受性の高い動物での; LD50の1/600以下、ED50の1/60以下、最大耐用量の1/60以下
(例)イヌでLD50=500mg/Kg の場合、60Kgのヒトでは、500*60/600=50mg となる。
B.薬物は5つの作用を持つ
興奮作用(stimulation)、抑制作用(depression)、刺激作用(irritation)、補充作用(replacement)、抗感染作用(anti-infective action)
C.薬物の相互作用
協力(synergism)→{ 相加(additive)、相乗(potentiation) }、拮抗(antagonism)
D.薬物の作用機序
薬物受容体(代謝系酵素、輸送蛋白質、チュブリン、核酸、イオンチャネル、細胞膜糖蛋白質、細胞膜脂質)
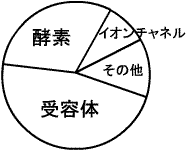
医薬品の作用点別の分布
作用薬(agonists)、部分作動薬(partial agonists)、拮抗薬(antagonists)、逆作動薬(inverse agonists)、競合的(competitive)拮抗薬、非競合的(noncompetitive)拮抗薬、薬物-受容体相互作用(質量作用の法則)、受容体の脱感作(homologous/heterologous desensitization)、細胞内情報伝達物質
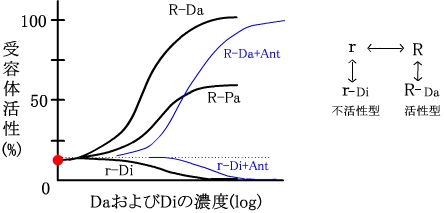
| 受容体は、不活性型(r)と活性型(R)の平衡状態にあり、リガンドがなくても若干が恒常的に活性化されている(●)。作動薬(Da)は活性型受容体(R)に親和性があり、平衡を活性型優位の方向にずらす。そして受容体は高濃度のDaで最大に活性化される。しかし、部分作動薬(Pa)では完全には活性化されない。逆作動薬(Di)は、不活性型受容体(r)に親和性があり、平衡を不活性型優位にずらす。そして、受容体の恒常的な活性化を抑制する。また、一定量の拮抗薬(Ant)存在下では、作動薬(DaとDi)の反応曲線は右方にシフトする(青線)。 |
E.薬物動態
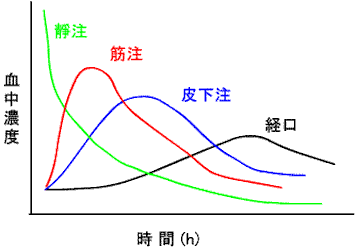
a.薬物の投与法:静脈、皮下、筋肉、舌下、吸入、経直腸、経口;
初回通過効果(first pass effect):経口および経直腸の一部から吸収した薬物は、肝臓を通り代謝され、体循環に入る。これを初回通過効果という。初回通過効果を受けやすい薬物として、ニトログリセリン(nitroglycerin)、プロプラノロール(propranolol)、レボドパ(levodopa)、イミプラミン(imipramine)などがある。
b.経口投与における、胃のpHによる薬物の解離度と薬物の吸収
pH=1.0の胃において、アスピリン(aspirin、pKa=3.5)では、非解離/解離=300/1であり、よく吸収される。ニコチン(nicotine、pKa=7.9)では、非解離/解離=1/1000000であり、ほとんど吸収されない。一般に、酸性物質は胃から吸収されやすく、塩基性物質は腸から吸収されやすい。
c.血液-組織関門:血液脳関門(Blood-Brain Barrier)
d.生体膜通過:拡散、輸送、小胞性輸送
e.血中薬物濃度とモデル
1)1分画モデル(one compartment model)
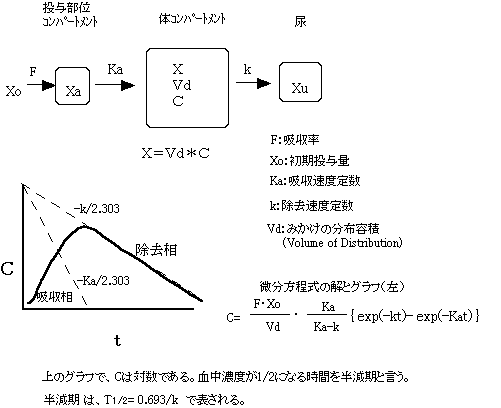
2)みかけの分布容積(Vd): 血管内5L、細胞間10L、細胞内20L:計35L
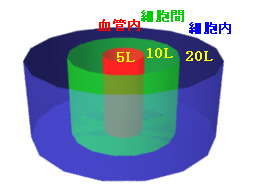
薬物が、血漿蛋白質と結合して血管内にととまればVdは5L以内、細胞間に分布すれば15L以内、細胞内に入れば35L以内、組織に蓄積すれば35L以上の値をとる。
3)体クリアランス(Clb):体内からの薬物の除去率の大きさを表す。
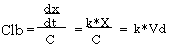
上の式から、Clbは単位時間にどれだけのVdを除去できるかを表す。
薬物の体クリアランスとみかけの分布容積
| Clb(L/h) | Vd(L) | 半減期(h) |
|---|---|---|---|
アスピリン(aspirin) | 39 | 11 | 0.25 |
クロロキン(chloroquine) | 45 | 13000 | 8.9 |
ジゴキシン(digoxin) | 7.8 | 440 | 39 |
イミプラミン(imipramine) | 63 | 1600 | 18 |
リチウム(lithium) | 1.5 | 55 | 22 |
ワルファリン(warfarin) | 0.2 | 9.8 | 37 |
腎臓から排泄される薬物と腎クリアランス
腎クリアランスの最大値は、腎血流量(650ml/min)に等しい39L/hである。 尿細管で再吸収も分泌もされない薬物(例えばイヌリン(inulin))のクリアランスは、糸球体濾過速度(125ml/min)に等しい7.5L/hである。腎クリアランスの最小値は、完全に再吸収される薬物(例えばglucose)で、0L/hである。
4)2分画モデル(two compartment model)註:以下の図の「抹消コンパートメント」は「末梢コンパートメント」の誤りです。
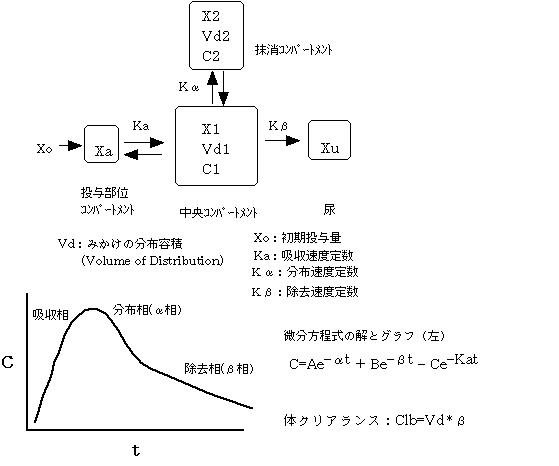
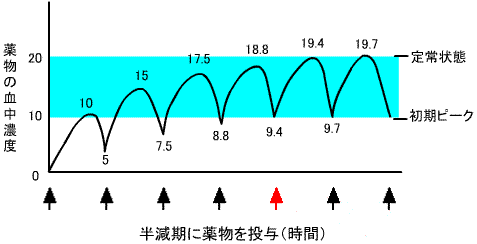
5)反復投与と薬物血中濃度
薬物を、静脈あるいは経口で反復投与する場合、投与間隔をその薬物の半減期にとると5回の投与により血中濃度は定常状態になり、血中濃度は初期ピークの2倍になる。点滴静注で持続的に投与する場合でも、その薬物の半減期の5倍の時間で定常状態になる。定常状態が有効血中濃度の範囲内にあることが重要である。もし、投与間隔を半減期の1/2にすると、初期ピークの3.4倍の血中濃度で定常状態になる。なお、薬物を間隔(τ)で投与した時の定常状態における平均血中濃(Cav)は、Cav=AUC/τで表される。
f.薬物の血漿蛋白質(albumin、β-globulinや酸性糖蛋白質)との結合
解離定数は Kd= 10-5-10-3
薬物の臨床効果発現は、フリー薬物血中濃度に比例することを基本としている。
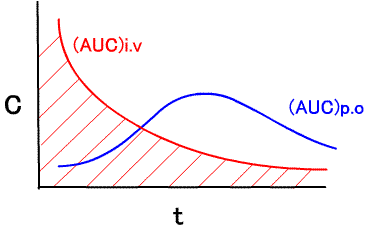
g.生体利用率(bioavailability)
AUC(area under curve):下図の赤線のカーブの下の面積
F=(AUC)p.o/(AUC)i.v ×100
F ≒ 経口投与した薬物の血中への移行量/全投与量 と考えられる。Fが大きいほど吸収がよく、投与量にみあった薬効が期待される。(参考)Vd=投与量/β/AUC でも求めることができる。生体利用率(F)は、薬剤の溶解性、粒子の性状、賦形剤や、食事や、消化管での吸収や肝臓での初回通過効果に依存している。相同面積の法則:等しい用量で完全な利用率の場合には、投与法に依存しない。薬剤の形を変更した時は、以前のものと生体利用率が同じかどうかを調べるために、同等試験を行う。
h.薬物の反復適応:蓄積、耐性、乱用、タキフィラキシー
i.血中薬物濃度のモニタリング:TDM(therapeutic drug monitoring)
安全域の狭い薬物は、適切な血中濃度を保つためにTDMが必要である。Vdの小さい薬物は、血漿蛋白質と結合しやすく、フリーの薬物濃度が変動しやすい。
F.薬物の代謝、排泄
薬物は一般的に脂溶性または非極性分子であり、細胞内に入りやすい。薬物排泄の基本は、薬物を酵素反応により極性増大の方向へ代謝し、水に溶けやすくして尿中に排泄する。
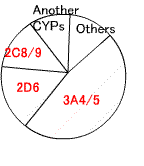
a.肝臓での代謝
第1相反応(酸化、還元、加水分解):-OH、-NH2を生じる。酸化反応は肝臓の滑面小胞体に局在する
P450(CPY)により行われる。
下図は、第1相酵素反応の割合(Goodman&Gilman's, 10版、2001より改変)。
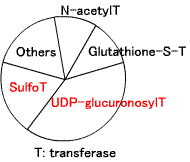
第2相反応(抱合、conjugation):水溶性原子団をつける。グルクロン酸基、硫酸基、アミノ酸(グリシン、グルタミン、オルニチン)、グルタチオン。
下図は、第2相酵素反応の割合(Goodman&Gilman's, 10版、2001より改変)
Cytochrome P450 (CYP)アイソザイムと代謝される薬
アイソザイム | 薬物 | その他 |
|---|---|---|
1A2 | カフェイン(caffeine), テオフィリン(theophylline), エストラジオール(estradiol) | chromosome 15 |
2C19 | プロトンポンプ阻害薬、 | mephenytoinの代謝活性低下(poor metabolizer)として見いだされ、白人3-5%、日本人15-20%に存在。 |
2C9 | NSAIDs、経口糖尿病治療薬、 | ワルファリン(warfarin)の代謝活性低下として見いだされた。 |
2D6
| β遮断薬、 抗うつ薬、 | debrisoquin, フェニトイン(phenytoin), フェナセチン(phenacetin)の代謝活性低下として見いだされ、日本人0.5%、白人5-10%に存在。 |
2E1 | 吸入麻酔薬、 | chromosome 10 |
3A4,5,7 | マクロライド系抗生物質、 | chromosome 7 |
b.腸肝循環
c.腎尿細管での排泄、再吸収
G.薬物相互作用
薬物療法は単独使用が基本である。これにより副作用の軽減と発見が容易になる。「配合剤」とよばれる、複数の薬成分を含む薬剤が多くなってきました(「配合剤のメリットとデメリット」はこちら)。
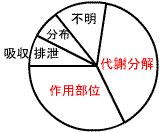
薬物相互作用が生じる部位の割合
a.薬物間の組み合わせ数
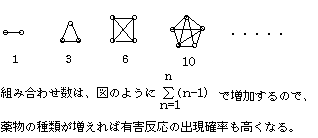
b、薬物動態力学的相互作用
| 相互作用の生じる場所 | 組み合わせ例と相互作用 | その他 |
|---|---|---|
| 吸収過程 | ・コレスチラミン(cholestyramine)(陰イオン交換樹脂、 胆汁酸吸着、高脂血症)により、酸性薬(ワルファリン(warfarin)、チアジド系、テトラサイクリン(tetracycline)、サイロキシン(thyroxine)、ジギタリス(digitalis))の吸収が阻害される。 | 吸着による吸収阻害 |
| ・消化管運動作用薬のコリン系とドパミン系薬物 | 吸収の促進や抑制 | |
| 分布過程 | ・アルブミンへの結合(ジギトキシン(digitoxin)、ワルファリン(warfarin)、ベンゾジアゼピン類など) ・α1酸性蛋白への結合(ジピリダモール(dipyridamole)、ジソピラミド(disopyramide)、キニジン(quinidine)、プロプラノロール(propranolol)など) | 血中の結合蛋白質に結合している薬物が、他の薬物と競合し、血中の遊離薬物濃度が上昇する。 |
| 代謝過程 | ||
| i)肝マイクロソームの薬物代謝酵素系(CYP)の誘導 | ・ワルファリン(warfarin)(フェノバルビタール(phenobarbital)によるCYP2C9の誘導により、血液凝固の延長) ・経口避妊薬(リファンピシン(rifampincin)によるCYP2C19誘導による避妊失敗) | 喫煙、バルビツレート(barbiturate)、リファンピシン(rifampincin)、抗てんかん薬などがCYPを誘導する。 |
| ii) CYP の阻害 | ・ケトコナゾール(ketoconazole)などの抗真菌薬によるCYP3A4阻害により、テルフェナジンの副作用の増強で、死に至る心室性不整脈が生じる。 ・ジヒドロピリジン系Caブロッカー(グレープフルーツの成分がCYP1A2を抑制し、作用増強)。 ・テオフィリン(theophylline)(ニューキノロンによるCYP1A2阻害による作用増強) | マクロライド系、キニジン(quinidine)、ニューキノロン類、サルファ剤、オメプラゾール(omeprazole)などが、CYPを阻害する。 |
| iii)CYP以外の代謝拮抗 | ・5-FU(抗腫瘍薬)の毒性がソリブジン(抗ウイルス薬)により増強される。 | ソリブジンの代謝物により、5-FU分解酵素(ジヒドロピリミジンデヒドロゲナーゼ)が阻害される。 |
| 排泄過程 | ||
| i)尿のpH変化による薬物再吸収の変動 | ・酸性尿(ビタミンC大量)で、酸性薬の排泄低下、塩基性薬の排泄増加 ・アルカリ尿(重曹、アセタゾラミド(acetazolamide))で、酸性薬の排泄増加、塩基性薬の排泄低下 | 酸性薬(アスピリン(aspirin)、フェノバルビタール(phenobarbital)、nalidixic acidなど) 塩基性薬(tetracyclin、イミプラミン(imipramine)、プロカイン(procaine)など) |
| ii)有機アニオン分泌系の競合 | ・プロベネシド(probenecid)による、酸性薬(ペニシリン類、インドメタシン(indomethacin)、ワルファリン(warfarin)、経口糖尿病薬)の分泌抑制により、薬物濃度の上昇 | |
| iii)有機カチオン分泌系の競合 | ・シメチジン(cimetidine)によるプロカインアミド(procainamide)の排泄抑制 | |
| iv)P-糖蛋白質の競合 | ・キニジン(quinidine)によるジゴキシン(digoxin)分泌抑制 ・イトラコナゾール(itraconazole)やベラパミル(verapamil)によるダビガトラン(dabigatran)血中濃度の上昇 | イトラコナゾールやベラパミルなどのP-糖蛋白質を強く阻害する医薬品を併用した場合、新規経口抗凝固薬のダビガトランのプロドラッグ体の吸収が亢進し、ダビガトランの血中濃度が上昇することが報告されており,抗凝固作用が増強され、大出血を起こす可能性がある。イトラコナゾールの経口製剤は,特に強い阻害作用を示すため,ダビガトランと併用禁忌となっている。 |
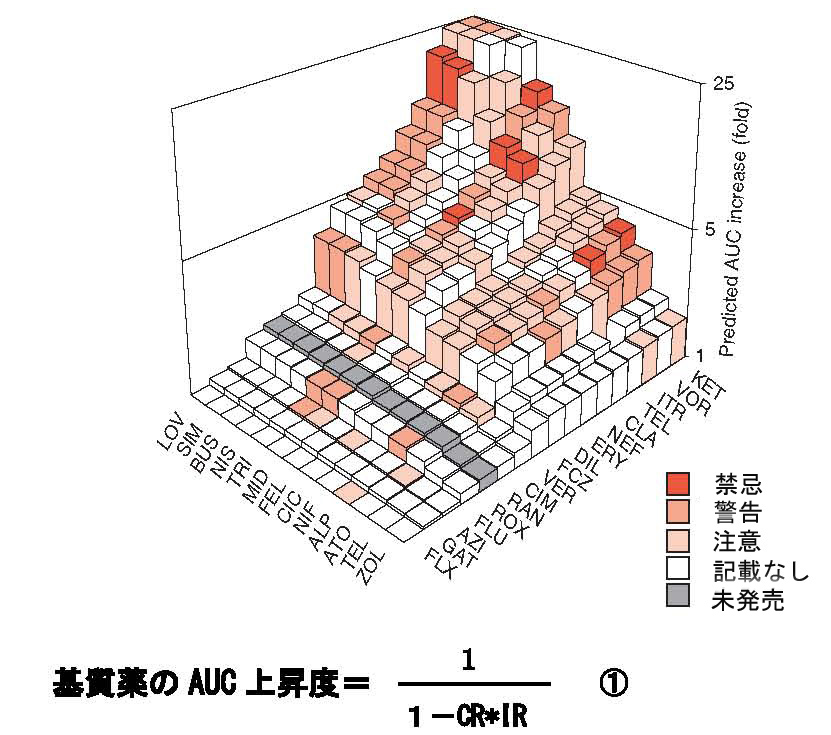
| CYP3A4の基質薬と阻害薬の相互作用による基質薬の血中濃度予測と、添付書類(米国)での警告 阻害薬(inhibitor)を併用したときの基質薬(substrate)のAUCを求めると、①式の関係が成立する。もしIR(inhibition ratio)とAUCが分かれば、CR(contribution ratio)が求められる。例えば、基質薬SIMを、阻害薬CLA(IRは0.88)と併用して、AUCを実測したとき、AUCの増加が8.3倍であれば、CRは1.0と計算される。上のグラフは、さまざまなCYP3A4の基質薬と阻害薬を併用したときのAUC増加倍数を予測したものである。添付書類(米国)で、薬物相互作用の警告が記載されている組み合わせは赤色の濃淡で示されいる。わが国の添付書類での警告記載は米国の58%である。 (図と説明は、A.Hisaka et al, Clin. Pharmacokinet. 48, 653, 2009より引用) 阻害薬(右から活性の強い順): ケトコナゾール(ketoconazole、KET), voriconazole (VOR), イトラコナゾール(itraconazole、ITR), telithromycin (TEL), クラリスロマイシン(clarithromycin、CLA), nefazodone (NEF), エリスロマイシン(erythromycin、ERY), ジルチアゼム(diltiazem、DIL), フルコナゾール(fluconazole、FCZ), ベラパミル(verapamil、VER), シメチジン(cimetidine、CIM), ラニチジン(ranitidine、RAN), roxithromycin (ROX), フルボキサミン(fluvoxamine、FLU), azithromycin (AZI), gatifloxacin (GAT) and fluoxetine (FLX). 基質薬(左から阻害薬に感受性の強い順): lovastatin (LOV), シンバスタチン(simvastatin、SIM), buspirone (BUS), nisoldipine (NIS), トリアゾラム(triazolam、TRI), midazolam (MID), felodipine (FEL), シクロスポリン(ciclosporin、CIC), ニフェジピン(nifedipine、NIF), alprazolam (ALP), アトルバスタチン(atorvastatin、ATO), telithromycin (TEL), and ゾルピデム(zolpidem、ZOL). |
c、薬力学的相互作用
| 作用の生じる部位 | 組み合わせ例と相互作用 | その他 |
|---|---|---|
| 同じ作用部位 (受容体など) | ・受容体刺激薬と遮断薬の併用 ・(隠れた)アゴニストとの併用 | ・気管支喘息のβ刺激薬と不整脈の作用 ・メトクロプラミド(metoclopramide)(消化器系機能改善薬)とチアプリド(tiapride)(脳循環改善薬)はともにD2受容体拮抗薬 |
| 異なる作用部位 (作用の収束) | ・気管支拡張薬(β刺激薬、methylxanthines、抗コリン薬(ipratropium)、抗ヒスタミン薬(第二世代)の併用による増強作用) ・抗凝固系薬(ワルファリン(warfarin)とアスピリン(aspirin)による抗凝固作用増強) ・血糖作用系の併用(スルホニル尿素類、β遮断薬、糖吸収遅延薬による低血糖の作用増強) |
H.薬物の有害反応
常用量の薬物の投与により、患者に起こるすべての望ましくない効果あるいは有害な効果をいう。
a.中毒反応
1)薬物特異的有害反応:一定以上の用量ならばだれでも起こりうる有害反応
2)宿主特異的有害反応
a)性、年齢、疾患、素因、遺伝などで、薬物の有害反応が異なる。
b)薬物代謝酵素欠損:薬物応答遺伝子多型によると考えられる。
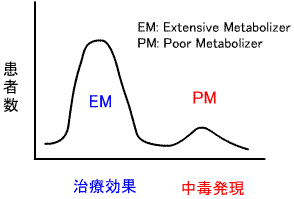
同一量を投与すると、薬物濃度の分布が2峰性となる。
肝薬物代謝酵素遺伝子欠損:イソニアジド(isoniazid)→末梢神経障害、クロラムフェニコール(chloramphenicol)→再生不良性貧血、エリスロマイシン(erythromycin)→肝障害
glucose-6-phosphate dehydrogenase欠損:sulfonamides, クロラムフェニコール(chloramphenicol)→溶血性貧血
b.アレルギー: I型(アナフィラキシー型)、II型(細胞障害型)、
III型(免疫複合型、アルザス型)、IV型(細胞仲介型)
c.二次的有害反応
d.催奇形成
多くの薬物は、胎盤を通過し、胎児に作用する。胎児への薬物移行は遅く、40分位で平衡状態になる。妊娠中の投薬で、妊娠と気づかないで投薬されたり、安全性の説明が十分でなかった場合が問題となる。
| 妊娠週齢 | 1~2 | 3~8 | 9~ |
|---|---|---|---|
| 器官形成 | 卵分割・着床 | 中枢神経、心臓、眼、上肢、下肢、耳、歯、 | 口蓋、外性器、脳 |
| 薬物感受性 | 薬物はほとんど影響しない | 絶対的過敏期である。 | 多くの薬物が発達障害を引き起こす。 |
| 注意すべき薬物 | 抗生物質、ホルモン剤、抗てんかん薬、抗不安薬、モルヒネ、トリアゾラム(triazolam)、methyldopa、ワルファリン(warfarin)、ACE阻害薬、コカイン(cocaine)、アルコール、リチウム | ホルモン剤、モルヒネ、トリアゾラム(triazolam)、methyldopa、ワルファリン(warfarin)、ACE阻害薬、コカイン(cocaine)、アルコール、抗うつ薬、 |
e.発癌性
近年、悪性腫瘍に対する治療の発達により生存率が上昇し生存期間も延長しているが、それとともに二次発がんの頻度も高くなることが長期疫学的調査により認識されている。そして二次発がんそのものが患者の生命予後を大きく左右することもわかってきた。米国でのデータによると、がん治療を受けた人の二次発がんのリスクは、がん治療を受けていない人に比べて25年間で14%高くなる。初発がんの発生年齢ごとにみると二次発がんのリスクは小児ほど高く、0-17歳では一般の人に比べ6倍、18-29歳で2.9倍、30-39歳で2.4倍、40-49歳で1.6倍、50-69歳で1.1-1.3倍であった。70歳以上では一般とほとんど変わらない。
関連サイトの紹介
1、MSDマニュアルプロフェッショナル版 薬物相互作用
2、医薬基盤・健康・栄養研究所 グレープフルーツと薬物の相互作用について
(三木、佐伯、久野)
